
Research
Reconstructing evolutionary history of cancer genomes
The reconstruction of cancer evolutionary history is important for early detection, prognosis, and treatment of patients. Phylogenetic inference methods have been widely used to reconstruct evolutionary history of cancer cells with different types of markers.
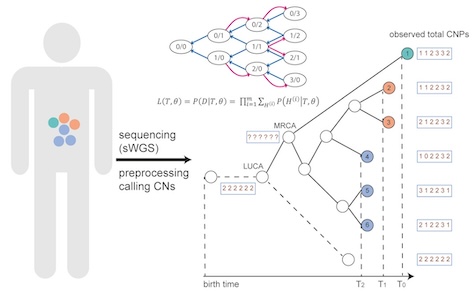
Using copy number alterations (CNAs) as markers, we have developed CNETML to jointly infer the tree topology, node ages, and CNA rates from longitudinal samples.
With the recent MRC award (2025 to 2028), we will develop more realistic models of evolution that integrate CNAs at different levels and Bayesian inference methods that support variable CNA rates in time.
References:
Bingxin Lu. (2024). Cancer phylogenetic inference using copy number alterations detected from DNA sequencing data. Cancer Pathogenesis and Therapy, 2, E27-E77.
Cheng Zhao, Darren P. Ennis, Bingxin Lu, Hasan B. Mirza, Chishimba Sokota, Baljeet Kaur, Naveena Singh et al. (2024). The genomic trajectory of ovarian high grade serous carcinoma can be observed in STIC lesions. The Journal of Pathology.
Bingxin Lu, Kit Curtius, Trevor A. Graham, Ziheng Yang, Chris P. Barnes (2023). CNETML: maximum likelihood inference of phylogeny from copy number profiles of multiple samples. Genome Biology, 24, 144.
Computational modelling of human genomes
Linking genomics with stochastic modelling and Bayesian inference provides a powerful approach to quantify somatic evolution, which may help to predict disease progression and drug response.
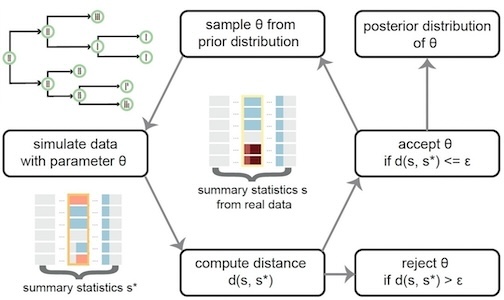
We have applied this approach to model CNAs and structural variants (SVs) resulted from chromosome instability from experimental and real cancer patient data.
Our previous inferences of important prognosis-related parameters including chromosome mis-segregation rates and selection strengths were limited by the mixture of signals in bulk sequencing data. Therefore, we are interested in improving the inference at a higher resolution with increasingly available single cell data.
We are also interested in applying the latest genomic language models to tackle challenges related to human genomics.
References:
Bingxin Lu, Samuel Winnall, William Cross, Chris P. Barnes (2025). Cell-cycle dependent DNA repair and replication unifies patterns of chromosome instability. Nature Communications, 16(3033).
William Cross*, Salpie Nowinski*, George Cresswell*, Maximilian Mossner*, Abhirup Banerjee*, Bingxin Lu*, et al. (2024). Negative selection may cause grossly altered but broadly stable karyotypes in metastatic colorectal cancer. bioRxiv. (* co-first authors)
Yannik Bollen, Ellen Stelloo, Petra van Leenen, Bas Ponsioen, Myrna van den Bos, Bingxin Lu, et al. (2021). Reconstructing single-cell karyotype alterations in colorectal cancer identifies punctuated and gradual diversification patterns. Nature Genetics, 53(8), 1187-1195.
Investigating intratumour heterogeneity and clonal evolution in cancer genomes
Intratumour heterogeneity (ITH) and clonal evolution often cause therapy failure and drug resistance in cancer patients. We worked on the analysis of genomic ITH in lung adenocarcinoma (LUAD) and hepatocellular carcinoma (HCC) patients previously.
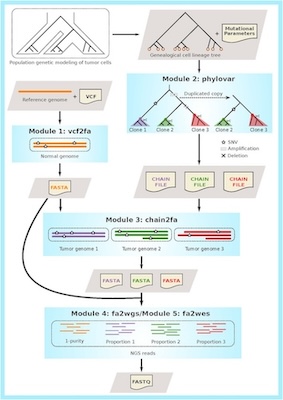
To evaluate the performances of different variant callers and clonal decomposition methods, we also developed a phylogeny guided simulator for tumour evolution (PSITE).
We are interested in developing new methods to decompose ITH and decipher clonal evolution.
References:
Kasper Karlsson, Moritz Przybilla, Eran Kotler, Aziz Khan, Hang Xu, Kremena Karagyozova, Alexandra Sockell, Wing H. Wong, Katherine Liu, Amanda Mah, Yuan-Hung Lo, Bingxin Lu, et al. (2023). Deterministic evolution and stringent selection during preneoplasia. Nature, 618, 383–393.
Jianbin Chen, Neslihan Arife Kaya, Ying Zhang, Raden Indah Kendarsari, Karthik Sekar, Shay Lee Chong, Veerabrahma Pratap Seshachalam, Wen Huan Ling, Cheryl Zi Jin Phua, Hannah Lai, Hechuan Yang, Bingxin Lu, et al. (2024). A multimodal atlas of hepatocellular carcinoma reveals convergent evolutionary paths and ‘bad apple’ effect on clinical trajectory. Journal of Hepatology, 4(81), 667-678.
Weiwei Zhai*, Hannah Lai*, Neslihan Arife Kaya*, Jianbin Chen*, Hechuan Yang*, Bingxin Lu*, et al. (2022). Dynamic phenotypic heterogeneity and the evolution of multiple RNA subtypes in Hepatocellular Carcinoma: the PLANET study. National Science Review, 9(3), nwab192. (* co-first authors)
Jianbin Chen, Hechuan Yang, Audrey Su Min Teo, Lidyana Bte Amer, Faranak Ghazi Sherbaf, Chu Quan Tan, Jacob Josiah Santiago Alvarez, Bingxin Lu, et al. (2020). Genomic landscape and ethnic specificity of lung adenocarcinoma in Asia. Nature Genetics, 52(2), 177-186.
Hechuan Yang, Bingxin Lu, Lan Huong Lai, Abner Herbert Lim, Jacob Josiah Santiago Alvarez, Weiwei Zhai (2019). PSiTE: A phylogeny guided simulator for tumor evolution. Bioinformatics, 35(17), 3148-3150.
Understanding the evolution of microbial genomes
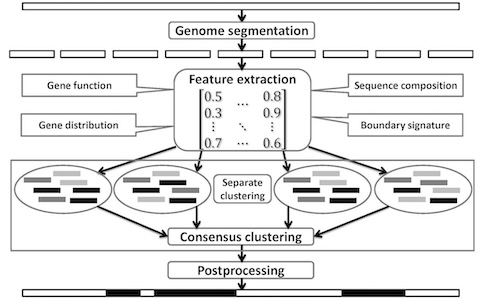
Lateral gene transfer (LGT) and recombination are common and important evolutionary processes in microbes. We have developed two machine learning methods to predict genomic islands (GIs), a large genomic region probably acquired by LGT which may contain genes related to pathogenesis and antibiotic resistance. We are also interested in developing and applying new methods to other important problems in microbial evolution.
References:
Bingxin Lu, Hon Wai Leong (2018). GI-Cluster: detecting genomic islands via consensus clustering on multiple features. Journal of Bioinformatics and Computational Biology, 16(03), 1840010.
Bingxin Lu, Hon Wai Leong (2016). Computational methods for predicting genomic islands in microbial genomes. Computational and Structural Biotechnology Journal, 14:200-206.
Bingxin Lu, Hon Wai Leong (2016). GI-SVM: A sensitive method for predicting genomic islands based on unannotated sequence of a single genome. Journal of Bioinformatics and Computational Biology, 14(01), 1640003.
Solving problems related to phylogenetic networks
Phylogenetic networks are becoming essential to represent complex evolutionary relationships when LGT and other reticulation events are involved.
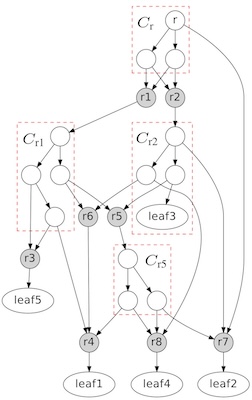
We previously developed algorithms related to two fundamental problems in phylogenetic networks, the tree containment problem (TCP) and the cluster containment problem (CCP). We are interested in solving other related problems.
References:
Andreas D.M. Gunawan, Bingxin Lu, Louxin Zhang (2018). Fast methods for solving the Cluster Containment Problem for phylogenetic networks. arXiv:1801.04498.
Bingxin Lu, Louxin Zhang, HonWai Leong (2017). A program to compute the soft Robinson–Foulds distance between phylogenetic networks. BMC Genomics, 18(2), 111.
Andreas D.M. Gunawan*, Bingxin Lu*, Louxin Zhang (2016). A program for verification of phylogenetic network models. Bioinformatics, 32(17), i503-i510. (* co-first authors)
Integrating multi-omics data to solve problems in cancer and microbial biology
To understand all the underlying processes shaping cancer evolution and inform treatment, it is necessary to integrate data measurements of various types. High-throughput sequencing has been generating huge amounts of multi-omics data, which provide a rich resource of information to address important questions in cancer evolution. However, it is challenging to systematically integrate these heterogeneous data types. Machine learning has emerged as a promising technique for multi-omics data integration, but there are still many challenges including data of high dimension yet low sample size, data noise and missing information, and biological interpretations. We are interested in developing new machine learning methods that leverage multi-omics data to tackle key challenges in cancer and microbial biology.
Despite multiple options and drugs to treat cancer, treatments often fail due to intratumour heterogeneity, metastasis, and drug resistance. However, it remains very expensive and time-consuming to develop new cancer drugs. Drug repurposing serves as a cost-effective option to provide patients with affordable and effective individualized treatments. We are interested in developing new machine learning methods for effective drug combinations or personalized drug recommendations.
References:
Jinwen Feng, Chen Ding, Naiqi Qiu, Xiaotian Ni, Dongdong Zhan, Wanlin Liu, Xia Xia, Peng Li, Bingxin Lu, et al. (2017). Firmiana: Towards a one-stop proteomic cloud platform for data processing and analysis. Nature Biotechnology, 35(5), 409-412.
Bingxin Lu, Zhenbing Zeng, Tieliu Shi (2013). Comparative study of de novo assembly and genome guided assembly strategies for transcriptome reconstruction based on RNA-Seq. Science China Life Sciences, 56(2), 143-155.
Reconstructing and analyzing genome graphs
SVs often alter large genomic regions and play an important role in both species evolution and cancer development. However, the complete landscape of SVs in the genomes has been understudied due to technical limitations and gradually gets improved with the new sequencing techniques such as long-read sequencing.
Due to the extreme variety of SVs, graph-based genome representation provides a natural way to analyze SVs, but the utilities of these graphs have not been fully exploited. We are interested in developing new approaches to better understand the patterns and mechanisms of SVs using genome graphs.